A doença de Gaucher é uma doença genética rara e hereditária, causada por uma deficiência da enzima beta-glicocerebrosidase. Essa enzima é responsável pela quebra de uma substância chamada glicocerebrosídeo, um tipo de gordura que se acumula nas células e em determinados órgãos quando a enzima está em falta ou funcionando inadequadamente. A acumulação de glicocerebrosídeo leva ao aumento do tamanho dos órgãos e ao comprometimento de sua função.
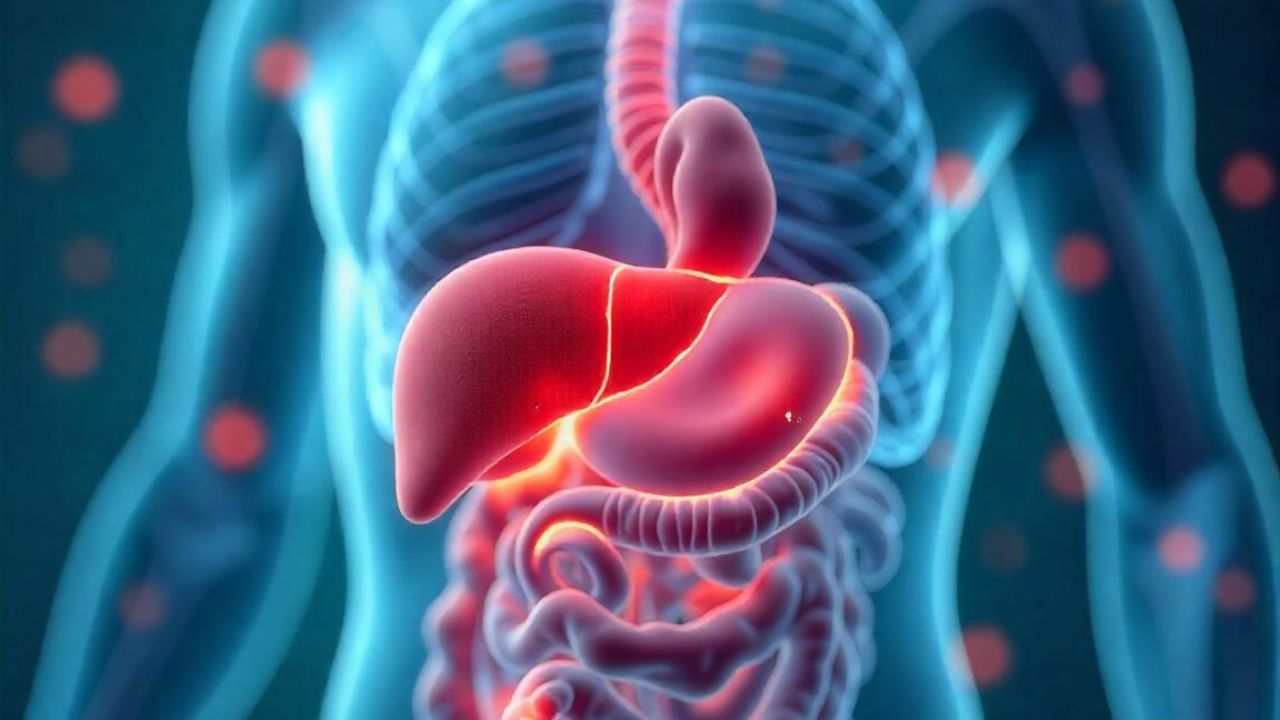
A doença de Gaucher é classificada em três tipos principais:
Tipo 1 (Forma Não Neurológica): É a forma mais comum e geralmente não afeta o sistema nervoso central. Afeta órgãos como baço, fígado e ossos, causando sintomas como aumento do baço e do fígado (esplenomegalia e hepatomegalia), anemia, fadiga, fraturas ósseas e dores ósseas.
Tipo 2 (Forma Neurológica Aguda): É a forma mais grave e afeta o sistema nervoso central. Os sintomas surgem geralmente nos primeiros anos de vida e incluem atraso no desenvolvimento, espasticidade, convulsões e comprometimento neurológico grave. Essa forma tem um prognóstico desfavorável e pode ser fatal na infância.
Tipo 3 (Forma Neurológica Crônica): É uma forma intermediária, com sintomas neurológicos mais leves que se desenvolvem de forma mais lenta. Os sintomas podem incluir problemas de movimento, convulsões, problemas oculares e sintomas semelhantes aos do Tipo 1, mas de forma progressiva.
Causas
A doença de Gaucher é causada por mutações no gene GBA, responsável pela produção da enzima beta-glicocerebrosidase. Quando o gene é defeituoso, a enzima não é produzida em quantidade suficiente ou é produzida de forma ineficaz, resultando no acúmulo de glicocerebrosídeo nas células.
Sintomas Comuns:
- Aumento do baço e fígado
- Dores ósseas e fraturas
- Anemia e fadiga
- Trombocitopenia (baixa contagem de plaquetas), levando a sangramentos e hematomas
- Em formas neurológicas, problemas de coordenação e desenvolvimento neurológico
Diagnóstico:
O diagnóstico da doença de Gaucher pode ser feito através de exames de sangue para medir a atividade enzimática da beta-glicocerebrosidase, testes genéticos para identificar mutações no gene GBA, além de exames de imagem para avaliar o tamanho do baço e do fígado e a condição dos ossos.
Tratamento:
O tratamento depende do tipo e da gravidade dos sintomas. As opções incluem:
Terapia de reposição enzimática (TRE): A administração regular da enzima faltante para compensar a deficiência e ajudar a reduzir o acúmulo de glicocerebrosídeo nas células.
Terapia de redução de substrato (TRS): Medicamentos que reduzem a produção de glicocerebrosídeo, diminuindo o acúmulo dessa substância nas células.
Tratamento de suporte: Inclui transfusões de sangue para anemia grave, analgésicos para dor óssea e intervenções ortopédicas para fraturas e outros problemas ósseos.
O acompanhamento contínuo com uma equipe médica especializada é essencial para o manejo adequado da doença e para melhorar a qualidade de vida dos pacientes com Gaucher.